Introduction: Many current therapies for autoimmune diseases such as multiple sclerosis (MS) act through nonspecific targeting of the immune response, rendering global immunosuppression, poor efficacy, and adverse side effects. To address a pressing need for safer and more effective therapies, multivalent soluble antigen arrays (SAgAs) have been developed to induce antigen-specific tolerance in MS. SAgAs consist of a flexible hyaluronic acid (HA) linear polymer cografted with multiple copies of autoantigen (proteolipid protein peptide, PLP139-151) and secondary inhibitory signal (ICAM-1 inhibitor peptide, LABL). These peptides are intended to target SAgA molecules to the immunological synapse, interrupting signaling between T cells and antigen presenting cells (APCs) to halt propagation of an autoimmune attack[1]-[4]. Previous in vivo studies showed SAgAs containing both signals were therapeutically effective against EAE (murine model of MS)[5]. We hypothesize SAgAs enact a therapeutic effect due to enhanced, antigen-specific binding with APCs. To investigate this hypothesis, in vitro studies were performed in a model APC system to investigate antigen-specific binding and receptor response; ex vivo studies were performed in murine splenocytes to investigate targeting of EAE immune cells.
Methods: 16 kDa HA was labeled with fluorescein isothiocyanate (FITC) to form ‘fHA’, then conjugated with aminooxy-LABL and/or -PLP to achieve a valency of ~10 peptides per HA backbone. Homopolymers (fHAPLP and fHALABL) were grafted with one peptide and a heteropolymer (fSAgAPLP:LABL) was co-grafted with both peptides. Conjugation was determined by RP-HPLC.
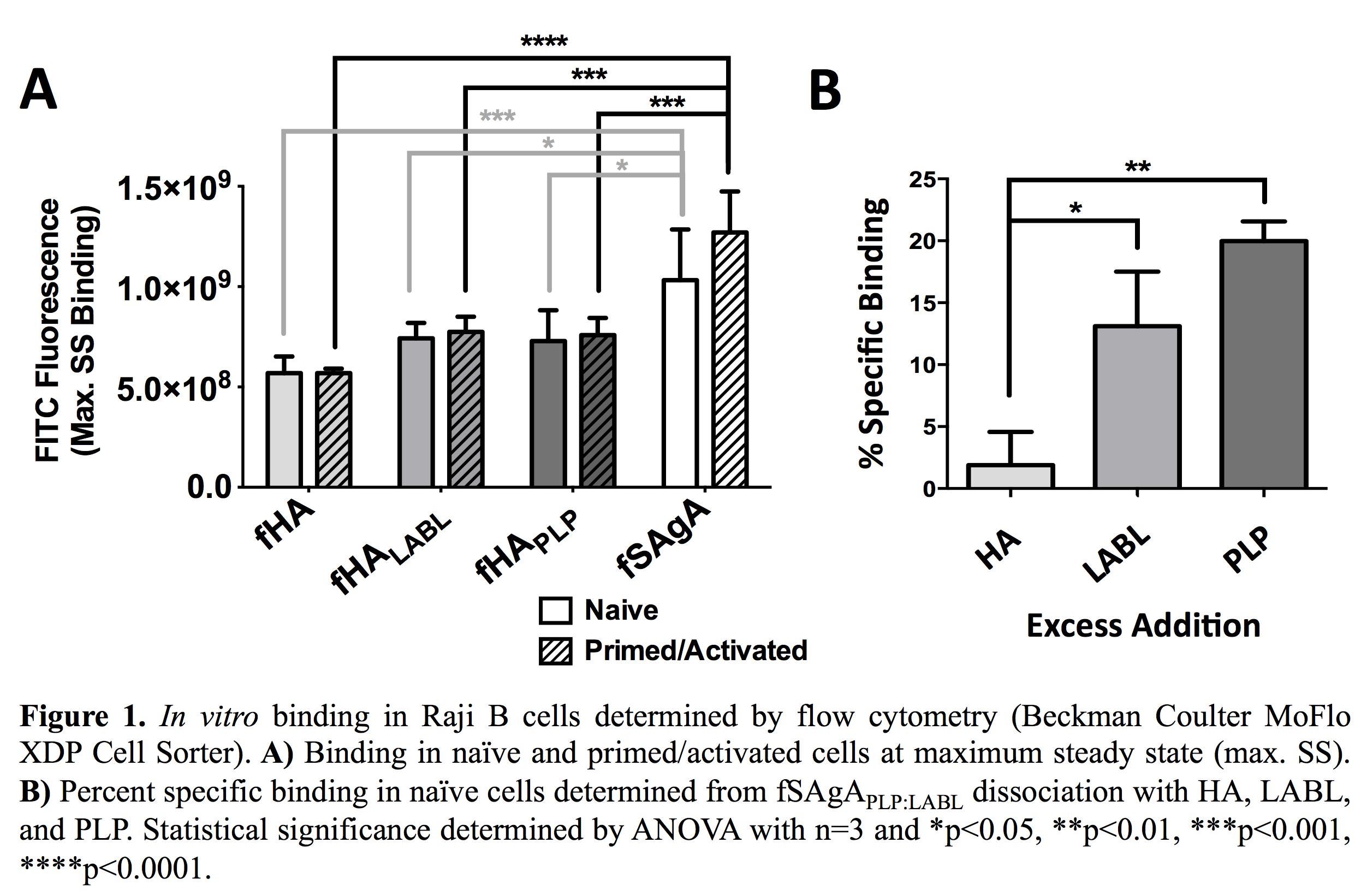
In vitro binding studies were performed in Raji B cells (APCs) using flow cytometry. Cells were primed and activated with TNF-α and PLP for 24 hours. To observe maximum binding, cells were mixed with labeled arrays immediately before injection on the cytometer and allowed to reach maximum steady state (SS). Specific binding was then determined through competitive dissociation with excess addition of unlabeled HA, PLP, or LABL. Cell binding and receptor clustering were observed in real-time by fluorescence microscopy using the CellASIC ONIX Microfluidics Platform and M04S switching plate.
Ex vivo binding studies were performed in splenocytes isolated from the spleens of both healthy and sick SJL/J mice, harvested at peak of disease following in vivo EAE induction. Isolated splenocytes were stained with CD3, CD22, and CD11c antibodies to identify T cells, B cells, and dendritic cells, respectively. Binding was evaluated by flow cytometry and fluorescence microscopy, as detailed above.
Results and Discussion: fSAgAPLP:LABL exhibited significantly greater binding in vitro than fHA, fHAPLP, and fHALABL. Specific binding determined by competitive dissociation was driven by PLP.
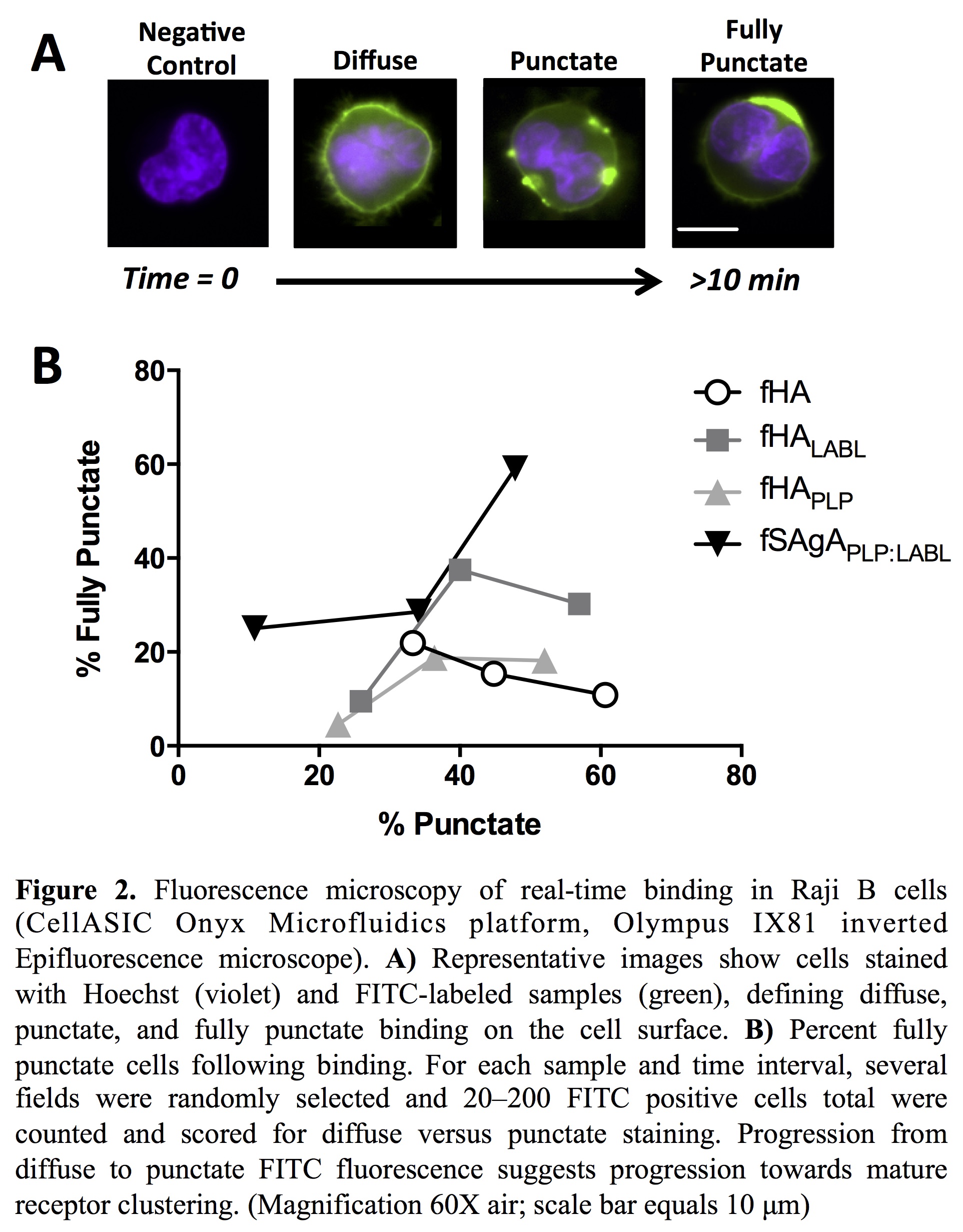
Receptor clustering, observed using real-time fluorescence microscopy, is an important precursor for signaling and activation of an antigen-specific cellular response. fSAgAPLP:LABL induced mature receptor clustering in Raji B cells to a greater extent than fHA, fHAPLP, or fHALABL.
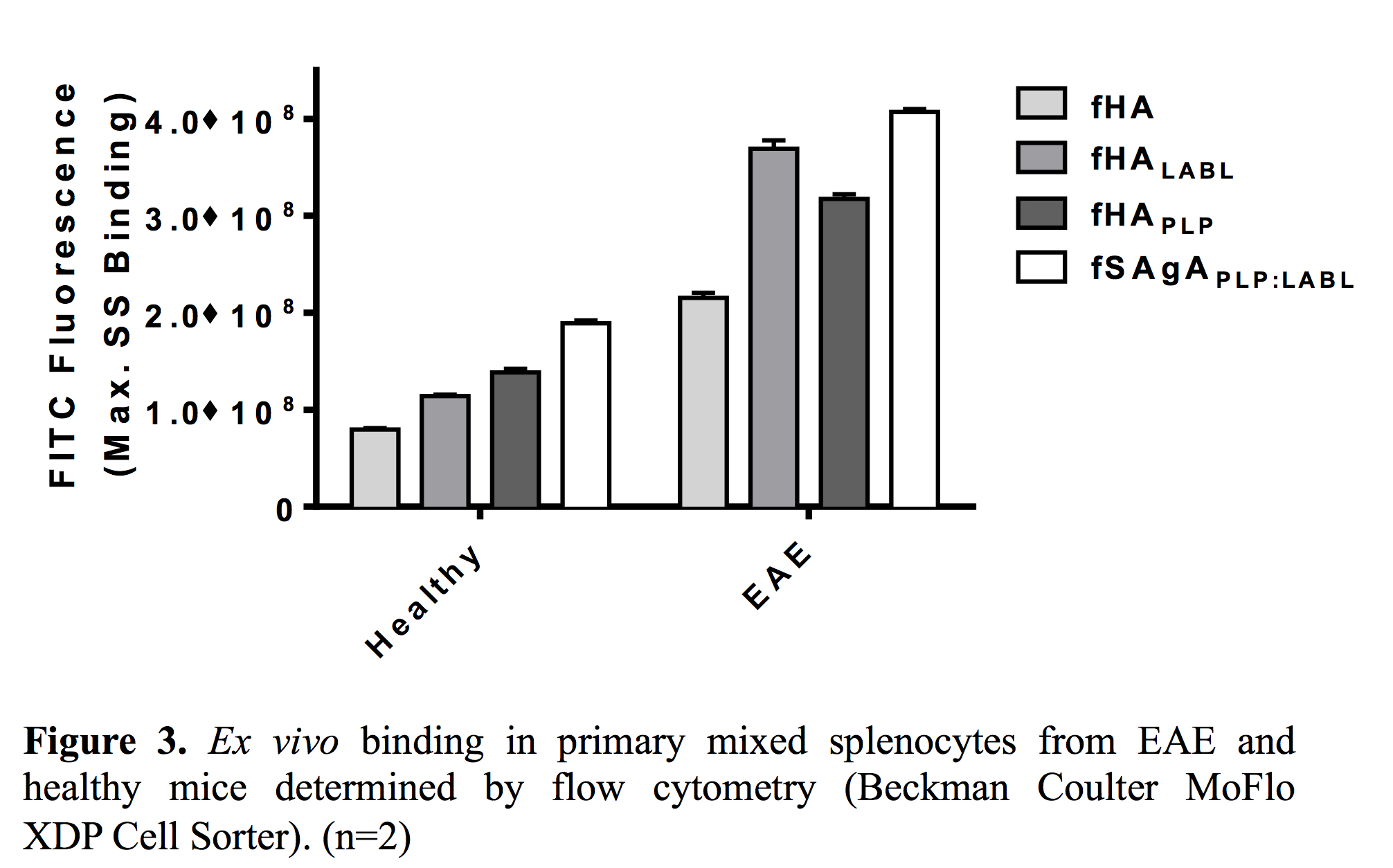
fSAgAPLP:LABL exhibited greater binding ex vivo than other arrays and greater binding in EAE splenocytes than healthy splenocytes.
Conclusions: SAgAPLP:LABL exhibited enhanced cell binding due to the presence of both PLP and LABL signals, primarily driven by the antigen. SAgAPLP:LABL binding was accompanied by progressive receptor clustering which may contribute to modulation of the immune response. Additionally, SAgAPLP:LABL exhibited greater binding with EAE splenocytes over healthy splenocytes. Leveraging enhanced, antigen-specific binding and receptor modulation, SAgAs offer a promising option for antigen-specific immunotherapy to repress autoimmune disease.
References:
[1] Grakoui, A.; Bromley, S. K.; Sumen, C.; Davis, M. M.; Shaw, A. S.; Allen, P. M.; Dustin, M. L., The immunological synapse: a molecular machine controlling T cell activation. Science 1999, 285 (5425), 221-227.
[2] Iezzi, G.; Karjalainen, K.; Lanzavecchia, A., The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 1998, 8 (1), 89-95.
[3] Baxter, A. G.; Hodgkin, P. D., Activation rules: the two-signal theories of immune activation. Nature Reviews Immunology 2002, 2 (6), 439-446.
[4] Bromley, S. K.; Iaboni, A.; Davis, S. J.; Whitty, A.; Green, J. M.; Shaw, A. S.; Weiss, A.; Dustin, M. L., The immunological synapse and CD28-CD80 interactions. Nature immunology 2001, 2 (12), 1159-1166.
[5] Sestak, J. O.; Sullivan, B. P.; Thati, S.; Northrup, L.; Hartwell, B.; Antunez, L.; Forrest, M. L.; Vines, C. M.; Siahaan, T. J.; Berkland, C., Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Molecular Therapy—Methods & Clinical Development 2014, 1.